Comprehensive Overview of Cystic Fibrosis (CF)
Introduction
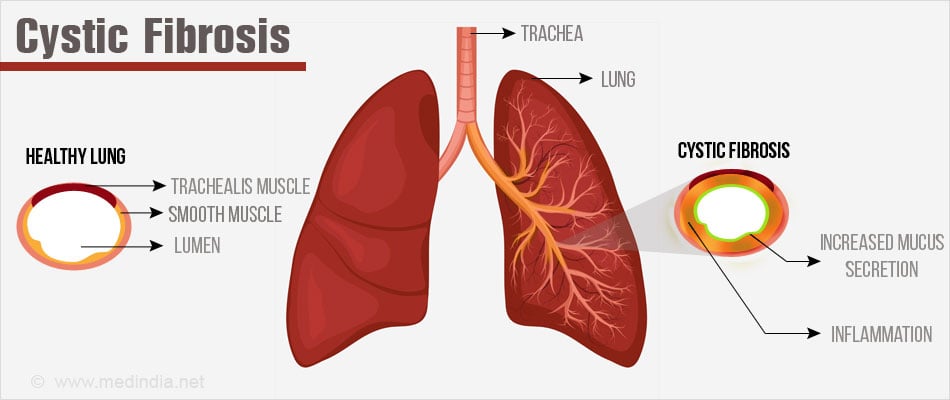
Cystic Fibrosis (CF) is a genetic disorder primarily affecting the lungs and digestive system. It's caused by mutations in the CFTR gene, leading to the production of thick, sticky mucus that clogs airways and ducts in various organs.
Epidemiology
- Prevalence: CF is the most common lethal inherited disease among Caucasians.
- Incidence: Approximately 1 in 2,500 newborns in the UK are affected, with a carrier rate of 1 in 25.
Pathophysiology

- CFTR Gene Mutation: Defects in this gene cause dysfunctional chloride channels, resulting in thick mucus secretions.
- Affected Systems: Primarily impacts respiratory, digestive, and reproductive systems due to mucus buildup.
Clinical Manifestations
Neonatal Period
- Meconium Ileus: Intestinal blockage is often the first sign.
- Prolonged Jaundice: May occur due to liver dysfunction.
Childhood
- Failure to Thrive: Caused by malnutrition due to malabsorption.
- Respiratory Symptoms: Chronic cough, recurrent lung infections, leading to bronchiectasis.
- Gastrointestinal Symptoms: Steatorrhea (fatty stools) and constipation.
Adolescence
- Respiratory Complications: Chronic infections, especially with Pseudomonas aeruginosa.
- Growth and Development: Delayed puberty and short stature.
Adulthood
- Diabetes Mellitus: Over 65% develop diabetes by age 25.
- Liver Disease: Fatty liver and gallstones are common.
- Infertility: Affects 98% of males due to vas deferens absence; females experience subfertility.
Diagnosis
Sweat Test
The primary diagnostic test for CF, measuring sweat chloride levels.
- Pilocarpine Iontophoresis: Stimulates sweat production using a mild electrical current.
- Collection: Sweat is collected over 30-60 minutes on filter paper or gauze.
- Analysis: Chloride concentration is measured.
Interpretation: >60 mmol/L chloride indicates CF; 40-60 mmol/L may require further testing; <40 mmol/L typically rules out CF.
Genetic Testing
Confirms CFTR mutations, essential for diagnosis, especially in borderline cases.
Common Infections
- Infants/Children: Staphylococcus aureus, Haemophilus influenzae.
- Adolescents/Adults: Pseudomonas aeruginosa, Burkholderia cepacia.
- Other Pathogens: Aspergillus, atypical mycobacteria.
Management
Multidisciplinary Approach
A collaborative effort involving various healthcare professionals.
- Respiratory Care: Regular chest physiotherapy, inhaled antibiotics, and DNase for mucus thinning.
- Nutritional Support: High-calorie diet, pancreatic enzyme supplements, and vitamins (A, D, E, K).
- Advanced Treatments: N-Acetylcysteine, lung transplantation, and investigational gene therapy.
Complications
- Cepacia Syndrome: Severe lung infection with Burkholderia cepacia.
- Reproductive Health: Male infertility, female subfertility due to mucus viscosity.
Prognosis
Median life expectancy is approximately 37 years, with improvements due to advanced care and therapies.
Additional Resources
- Cystic Fibrosis Foundation: Offers support and information for patients and families.
- Clinical Trials and Research: Ongoing studies on new treatments and gene therapy.
- Genetic Counseling: Important for families with a history of CF, available through healthcare providers.
Conclusion
Cystic Fibrosis requires a comprehensive, multidisciplinary approach to manage its complex symptoms and improve patient outcomes. Advances in treatment and ongoing research provide hope for further improvements in quality of life and survival rates.
Post a Comment